Here are some of the components of the common subgraph. As described in the paper, they span a variety of different topologies. The nodes in the graphs below are identified by the number of the corresponding orthlolog set (described later in this page):
Tree:
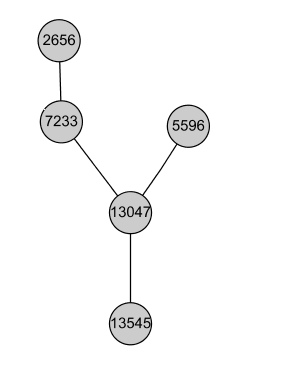
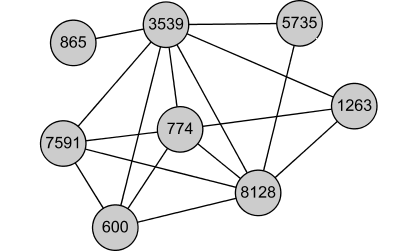
Complex. Also, many of the genes perform similar function: they are known to be part of the RNAase complex:
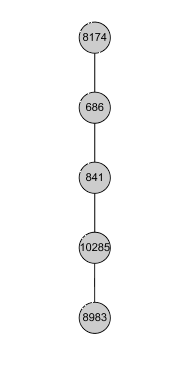
Linear Pathway:
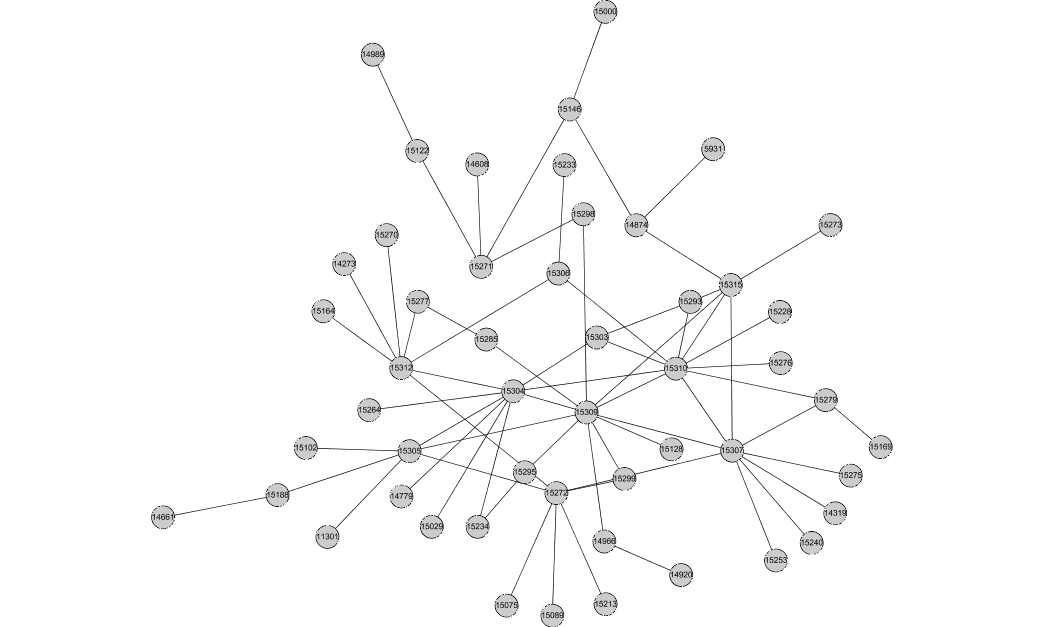
This is the largest connected component consisting of edges conserved in 3 or more species:
Here is the Cytoscape .sif file corresponding to the entire common subgraph.
gene-name cluster-idx
All the genes with the same cluster-idx are functional orthologs, i.e., they are mapped to each other. The gene names were custom-created for our purposes (because we integrated data from multiple databases). In each gene's name, the first 2 letters of the name indicate its species: e.g., "dm123" is a fly gene,d "sc1231" is a yeast gene, "hs2312" is a human gene etc. The translation of these names to common names (from Ensembl etc.) is here. Each line in this file contains a custom gene-name and the list of common synonyms for it.
We describe here our algorithm for computing functional coherence of an ortholog list:
- Given an ortholog list, identify each set
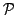 of orthologous proteins that has at least a fraction
of orthologous proteins that has at least a fraction 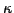 of its proteins with at least
one GO term. In our experiments, we chose
of its proteins with at least
one GO term. In our experiments, we chose 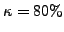 . Also, while
counting GO terms, we ignored those that had the IEA, ISS, or ND tags,
i.e., terms that had been assigned by electronic annotation, sequence
homology or without documented evidence. For each such set compute a
functional coherence score:
. Also, while
counting GO terms, we ignored those that had the IEA, ISS, or ND tags,
i.e., terms that had been assigned by electronic annotation, sequence
homology or without documented evidence. For each such set compute a
functional coherence score:
- Collect all the GO terms corresponding to any protein in (except those with ISS, IEA or ND tags).
- Map each GO term to one or more of a standardized set of GO terms.
In our case, this standardized set
consists of
GO terms that are at a distance 5 from the root of the GO tree. Each GO
term
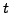 is mapped to its ancestor(s) at this level. If itself is
higher than level 5 in the GO hierarchy, it is removed from the
collection (this also helps remove functional annotation that is not specific enough). The intuition behind mapping GO terms to such a standardized
set is to avoid problems caused by measuring similarity between terms
at different levels of the GO hierarchy.
is mapped to its ancestor(s) at this level. If itself is
higher than level 5 in the GO hierarchy, it is removed from the
collection (this also helps remove functional annotation that is not specific enough). The intuition behind mapping GO terms to such a standardized
set is to avoid problems caused by measuring similarity between terms
at different levels of the GO hierarchy.
- Compute the similarity between every pair of standardized GO terms in .
Given terms
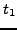 and
and 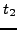 , let
, let 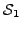 and
and 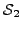 be the sets of proteins (across all input species) that have been assigned
GO terms that map to and , respectively. We define the
similarity between the two terms as:
be the sets of proteins (across all input species) that have been assigned
GO terms that map to and , respectively. We define the
similarity between the two terms as:
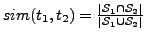 . The
. The 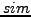 measure ranges from 0 (no overlap) to 1
(exact overlap).
measure ranges from 0 (no overlap) to 1
(exact overlap).
- Let
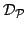 be the set of all pairwise distances between GO terms in . We define the functional coherence score of
be the set of all pairwise distances between GO terms in . We define the functional coherence score of
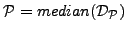 .
.
- Collect all the GO terms corresponding to any protein in
- Compute the average of the functional coherence scores over all the chosen sets.
We used Inparanoid, a popularly used orthology mapping program to estimate if the functional coherence scores are biologically relevant. Our intuition was that Inparanoid produces orthology mappings between pairs of species. By our knowledge of relative evolutionary distances between species, we can make reasonable guesses about the relative quality of these orthology mappings. Then, our functional coherence scores are plausible if they reflect the relative difference in quality between the various Inparanoid orthology lists. Here are the functional coherence scores as calculated for some of the orthology mappings produced by Inparanoid:
| Species 1 | Species 2 | Func. Coherence Score |
|---|---|---|
| Human | Mouse | 0.233 |
| Human | Yeast | 0.224 |
| Mouse | Yeast | 0.223 |
| Worm | Fly | 0.174 |
| Worm | Mouse | 0.139 |
| Worm | Human | 0.162 |
In the first block, the functional coherence (F.C.) scores of the pairwise orthology mappings of three species (Human, Mouse, and Yeast) are shown. Since human and mouse are evolutionarily much closer to each other than to yeast, it is to be expected that Inparanoid's human-mouse ortholog mapping will be better at grouping genes of shared origin (and shared function). This is borne out by the scores: the human-mouse F.C. scores are significantly higher than the human-yeast and mouse-yeast F.C. scores.
Similarly, in the second block we show F.C. scores for Inparanoid's orthology mapping between worm and three other species: human, mouse and fly. The F.C. scores indicate that worm-fly mapping is of higher quality than the worm-human and worm-mouse mapping. This is also plausible: it has been suggested (based on phylogenetic analysis) that the worm is closer to the fly than to mouse (or human) [1, 2]
The eigenvalue problems constructed in our algorithm are similar to those constructed in Google's PageRank algorithm. The intuition behind these constructions have a similar flavor. In PageRank, a web-page is ranked highly if it is pointed to by other web-pages of high rank; in our approach, a pair of proteins have a high score if their pairings of their respective neighbors have a high score. The actual algorithms in the two cases are quite different--- PageRank tries to rank nodes (i.e. web-pages) in a single graph, while our goal is to match nodes across multiple networks.