Current release: MixMapper v2.0 (May 29, 2014)
The MixMapper software performs admixture inference from allele frequency moment statistics as described in:
-
Original MixMapper paper:
Lipson M, Loh P-R, Levin A, Reich D, Patterson N, and Berger B. Efficient moment-based inference of population admixture parameters and sources of gene flow. Molecular Biology and Evolution, 2013. -
MixMapper 2.0:
Lipson M, Loh P-R, Patterson N, Moorjani P, Ko Y-C, Stoneking M, Berger B, and Reich D. Reconstructing Austronesian population history in Island Southeast Asia. bioRxiv, 2014 (under revision).
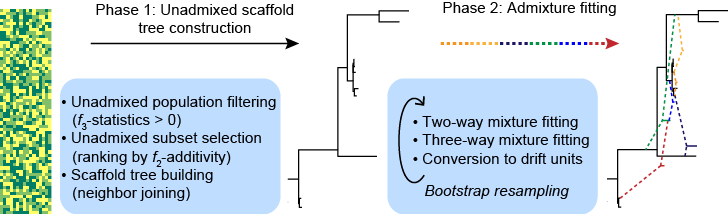
Abstract: The recent explosion in available genetic data has led to significant advances in understanding the demographic histories of and relationships among human populations. It is still a challenge, however, to infer reliable parameter values for complicated models involving many populations. Here we present MixMapper, an efficient, interactive method for constructing phylogenetic trees including admixture events using single nucleotide polymorphism (SNP) genotype data. MixMapper implements a novel two-phase approach to admixture inference using moment statistics, first building an unadmixed scaffold tree and then adding admixed populations by solving systems of equations that express allele frequency divergences in terms of mixture parameters. Importantly, all features of the tree, including topology, sources of gene flow, branch lengths, and mixture proportions, are optimized automatically from the data and include estimates of statistical uncertainty. MixMapper also uses a new method to express branch lengths in easily interpretable drift units. We apply MixMapper to recently published data for HGDP individuals genotyped on a SNP array designed especially for use in population genetics studies, obtaining confident results for 30 populations, 20 of them admixed. Notably, we confirm a signal of ancient admixture in European populations---including previously undetected admixture in Sardinians and Basques---involving a proportion of 20-40% ancient northern Eurasian ancestry.
Related Work
MixMapper can be thought of as a generalization of the qpgraph software of Patterson et al. (Genetics, 2012), which takes as input genotype data, along with a proposed arrangement of admixed and unadmixed populations, and returns branch lengths and mixture fractions that produce the best fit to allele frequency moment statistics measured on the data. MixMapper, by contrast, performs the fitting in two stages, first constructing an unadmixed scaffold tree via neighbor-joining and then automatically optimizing the placement of admixed populations onto this initial tree. Thus, no topological relationships among populations need to be specified in advance.
MixMapper is also similar in spirit to the independently developed TreeMix method of Pickrell et al. (PLoS Genetics, 2012). Like MixMapper, TreeMix builds admixture trees from second moments of allele frequency divergences, although it does so via a composite likelihood maximization approach made tractable with a multivariate normal approximation. Procedurally, TreeMix is structured in a "top-down" fashion, whereby a full set of populations is initially fit as an unadmixed tree, and gene flow edges are added sequentially to account for the greatest errors in the fit. This format makes TreeMix well-suited to handling very large trees: the entire fitting process is automated and can include arbitrarily many admixture events simultaneously. In contrast, MixMapper is designed as an interactive tool to maximize flexibility and precision with a "bottom-up" approach, beginning with a carefully screened unadmixed scaffold tree to which admixed populations are added with best-fitting parameter solutions.
Source Code and Executable (updated 5/29/14)
Our source code, written in C++ and MATLAB, is available for academic and non-profit use. Additionally, for users who do not have access to MATLAB, we provide a compiled executable that can be run with an install of the (free) MATLAB Compiler Runtime. (The MCR version is more cumbersome to use, however.)
The package contains a detailed README explaining how to install and use the software. Note that the MATLAB code for mixture fitting requires a MATLAB installation with the Bioinformatics Toolbox and Optimization Toolbox. If the Parallel Computing Toolbox is also installed, fitting of bootstrap replicates can be performed in parallel. We have tested the code on MATLAB versions 7.14 (R2012a) and 8.0 (R2012b).
Change Log
-
Version 2.0 (May 29, 2014):
-
Implemented new features for improved three-way admixture inference:
- More accurate 3-way mixture fraction inference
- Option for comparing goodness of fit of 2-way vs. 3-way models
- Fixed minor bug that caused a crash in rare cases (reroot_dist < 0).
- Fixed error in example data (h_boots file, which had been generated using outdated code).
-
Implemented new features for improved three-way admixture inference:
-
Version 1.02 (Aug 9, 2013): MixMapper_v1.02.tar.gz, MixMapperMCRdistrib_v1.02.tar.gz
- Compiled MixMapper into an executable now available for use with the MATLAB Compiler Runtime.
- Fixed bug in compute_most_additive_trees.cpp that caused crashes in some cases when not requiring any populations to be in the tree or when analyzing greater than 32 putatively unadmixed populations. The program now supports up to 64 unadmixed populations as intended.
- Fixed minor bug in MixMapper.m that caused error when options.branch_sets was not set.
- Updated compute_moment_stats.cpp to ignore any individual with "ignore" or "Ignore" in the population name.
-
Version 1.01 (Apr 11, 2013): MixMapper_v1.01.tar.gz
- Fixed bug in compute_moment_stats.cpp heterozygosity computation (missing factor of 2). This bug caused errors in the conversion of branch lengths to drift units; it did not affect the results when displayed in the default f2 units and did not affect the fitting procedure in either case.
- Fixed minor bug affecting root placement in rare cases.
- Oriented MixMapper output so that when displaying a mixture fit between Branch 1 and Branch 2, the major component of ancestry always comes from Branch 1 (i.e., the mixture fraction alpha > 0.5).
- Added option to name branches according to the sets of populations they split the tree into (as an alternative to the current "trace" naming system) and added output fields set1, set2, set3. Enable the 'branch_sets' option to display output using this nomenclature.
- Added support for input in "packed geno" format (2 bits/genotype).
-
Version 1.0 (Dec 3, 2012): MixMapper_v1.0.tar.gz
- Initial release.
Contact
We welcome feedback, questions and suggestions. Contact information is available at the primary authors' websites:
- Mark Lipson, Po-Ru Loh, Bonnie Berger