The explosive growth of single-cell transcriptomics (scRNA-seq) has transformed research on the regulation of cell differentiation, enabling an unprecedented reconstruction of cell lineages from which causal transcription factors (TFs) can be hypothesized. However, the bottleneck has now shifted to the validation of these hypotheses. Since most scRNA-seq studies are observational, with lineage-determining TFs predicted by correlational analyses, perturbational experiments are required to test these predictions and confirm causality. Yet such validations remain slow and expensive and are far outpaced by high-throughput scRNA-seq assays. To optimize limited experimental resources, computational approaches are needed to identify high-confidence scRNA-seq hypotheses and efficiently guide in vivo validations.
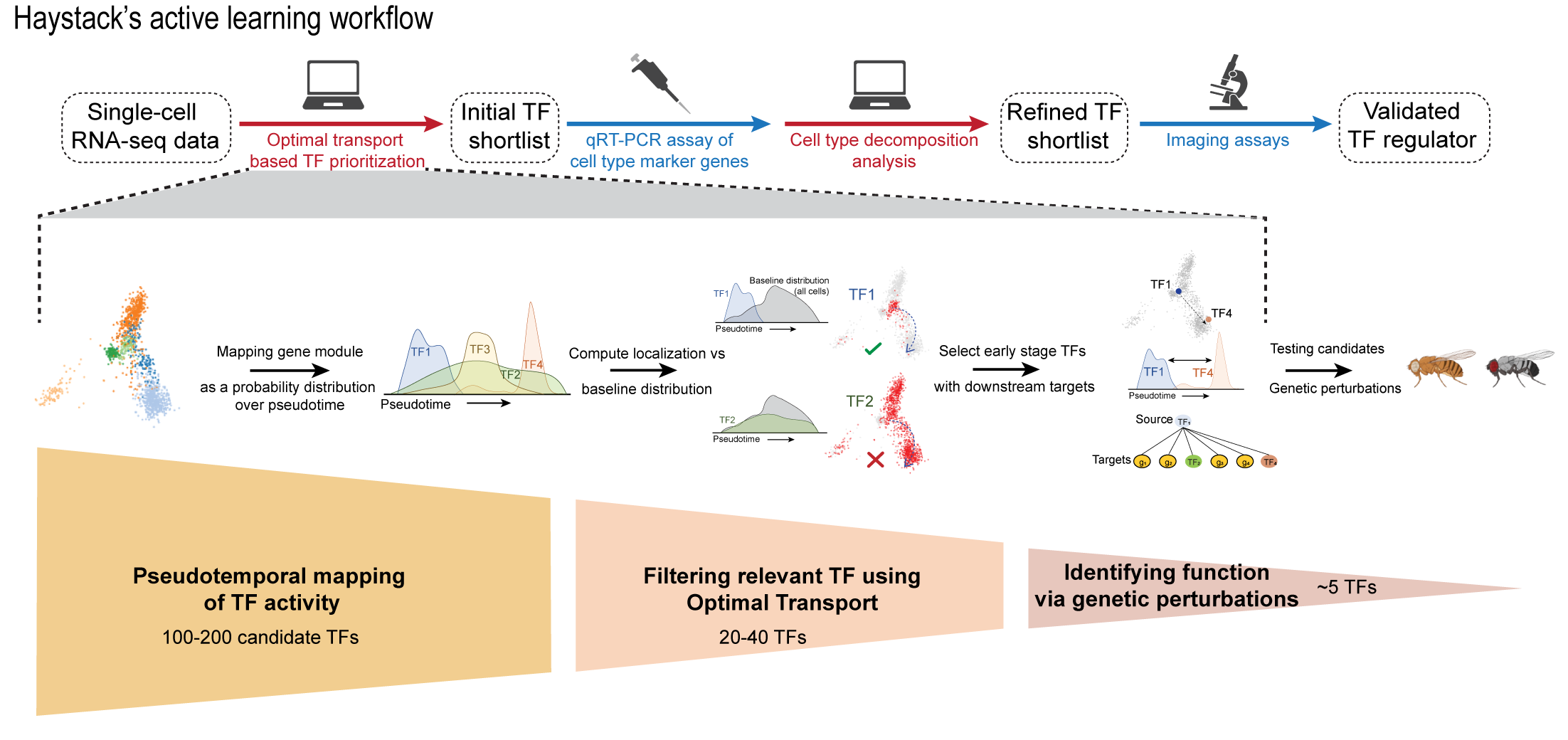
We present a hybrid computational-experimental method, Haystack, to identify TFs involved in differentiation from scRNA-seq datasets. TFs are computationally prioritized using an optimal transport formulation and then validated in an active learning framework that iteratively prunes validation targets. We offer Python code (below) for the computational shortlisting of TFs via optimal transport.
Haystack is described in the bioRxiv preprint, Optimal transport analysis of single-cell transcriptomics directs hypotheses prioritization and validation by Rohit Singh*, Joshua Li*, Sudhir Tattikota*, Yifang Liu, Jun Xu, Yanhui Hu, Norbert Perrimon, and Bonnie Berger.