 |
Carnelian: enhanced metabolic functional profiling of whole metagenome sequencing reads |
|
Massachusetts Institute of Technology (mit) AboutWe present Carnelian, a pipeline for alignment-free functional binning and abundance estimation, that leverages low-density even-coverage locality sensititve hashing to represent metagenomic reads in a low-dimensional manifold. When coupled with one-against-all classifiers, our tool bins whole metagenomic sequencing reads by molecular function encoded in their gene content at significantly higher accuracy than existing methods, especially for novel proteins. Carnelian is robust against study bias and produces highly informative functional profiles of metagenomic samples from disease studies as well as environmental studies. This work is presented in the following paper:
Sumaiya Nazeen, Yun William Yu, and Bonnie Berger*. Carnelian uncovers hidden functional patterns across diverse study populations from whole metagenome sequencing reads. Genome Biology 2020, 21(1): 47. [Link to paper] Availability
Note on Availability of Data Sets from Alm Lab:
Docker containerA docker container for Carnelian is available with all its dependencies. Please follow the following instructions.
Note on Requirements: Carnelian was tested with GCC 6.3.0 on Ubuntu 17.04, running under Bash 4.4.7(1) on a server with Intel Xeon E5-2695 v2 x86_64 2.40 GHz processor and 320 GB RAM. If you are planning to run it on a laptop/macbook, be sure to adjust the "--bits" parameter during training according to the available memory on your machine. Use a value as large as possible for the "--bits" parameter (default: 31; you might need to lower it if your RAM is smaller in size). Workflow
Helper scripts for analyses performed in the paper:
Pre-trained EC and COG models:All models are trained with vowpal-wabbit 8.1.1 and will not work with other versions of vowpal-wabbit. See full requirements on github.
Performance comparison between default mode and precise mode of Carnelian:Benchmarks were performed on subsets of EC-2010-DB data set. For the seen protein case, 100 bp reads were randomly drawn from the backtranslated protein sequences. To simulated mutated proteins, 5% mutation was randomly introduced in the reads. For hold-out experiment, proteins were randomly held out from multi-protein EC bins in our gold standard database. 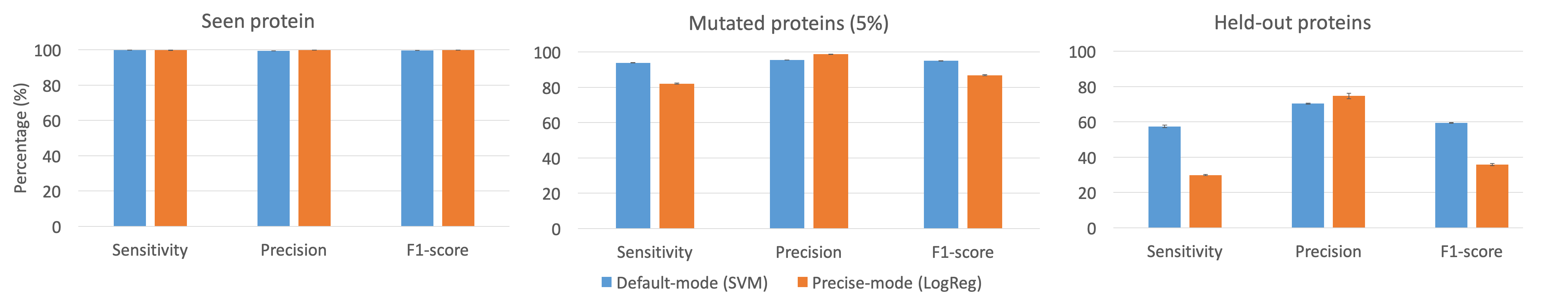 We recommend using Carnelian in default mode as it has been thoroughly tested. The purpose of precise mode is only to provide probability scores for the EC labels and this mode is suitable for smaller databases. Supplementary InformationMetadata associated with the real data sets analyzed in our study can be found here. © All rights reserved to Sumaiya Nazeen. 2018. This work is published under MIT License. Permission is hereby granted to whoever obtains a copy of the files associated with our pipeline to use, copy, modify, merge, publish, and distribute free of charge. For details see the license file. |