Index
About BetaWrapPro
What it does:
BetaWrapPro is a program for structural motif recognition and comparative modeling of the
pectin lyase-like single-stranded right-handed parallel beta-helix and the beta-trefoil
structural motifs (henceforth referred to as the beta-helix and beta-trefoil motifs for brevity).
It incorporates structural information about the folds, statistical residue
pair preferences in beta-sheets, and evolutionary information in the form of
sequence profiles to find potential alignments of the sequence to an abstract
structural template.
These alignments are then used with a known backbone and SCWRL, a sidechain packing program, to create
a 3D structural model of the predicted fold, which can then be viewed and
manipulated in the same way as any other PDB file.
Because the model is not created by sequence alignment but by pairwise residue
probabilities, BetaWrapPro is able to successfully model
proteins with very low sequence similarity to the backbone used for modeling.
What it doesn't do:
BetaWrapPro uses very little information about the sequences
of known beta-helices and beta-trefoils. It does not compare a sequence to
the known beta-helices and beta-trefoils, or any other known structures,
except to build a sequence profile (you do have the option of running an
additional search against Pfam HMMs, see below.
You should definately do these comparisons for any sequences you are
interested in, using for example the NCBI's BLAST service. BetaWrap is not a
threading program per se, in that it doesn't compare the sequence to any
other possible template structures. As a result it is much faster than
threading programs, but it won't notice if your sequence, which might make a
mediocre beta-helix, would in fact make a fantastic transmembrane beta-barrel.
You should consider using some version of threading or profile program for
sequences picked out by BetaWrap (for example 3D-PSSM). But don't be concerned
if threading doesn't support BetaWrap's prediction (as long as it doesn't find
a highly significant alternative hit) -- the threading programs we tried did
not do so well in recognizing similarity between many of the known
beta-helices.
How Well It Works
Motif recognition:
BetaWrapPro is able to predict the beta-helix motif with 100% sensitivity at 99.5% selectivity, and the beta-trefoil with 100% sensitivity at 92.5% selectivity in our experiments.
Sequence-structure alignment:
It produces sequence-structure alignments that are 76% accurate on the known beta-helix structures, and 86% on the beta-trefoils.
We note that it seems to perform less well on the pectin methylesterase SCOP family of beta-helices, and the beta-helix alignments of all structures not in this family are 84% accurate overall.
We define an alignment to be accurate if it is within three residues of the exact position in the solved structure.
Structure Prediction:
The accurately aligned regions of the predicted beta-helices average less than 2.0 Å RMSD (root mean square deviation) from the solved structures. The sidechain χ1 angles are correct for 61% of these residues, and χ1+2 are correct for 39%.
For the beta-trefoils, the backbone RMSD averages 4.5 Å, with 49% of χ1 angles correct and 25% of χ1+2.
It is likely that the higher RMSD for the trefoils compared to the helices is due to the greater structural deviaton between families of trefoils predicted by
BetaWrapPro, which in turn makes sidechain prediction more difficult.
An angle is counted as correct if it is within 40° of the angle in the solved structure.
Interpreting Your Results
Statistical significance:
Each sequence is assigned a raw score by the BetaWrap algorithm. This score
measures the compatibility of the sequence with a beta-helical structure. A
P-value is attached to this score which gives a rough estimate of the
likelihood that a randomly chosen sequence from the PDB that does not form the
template fold would attain a similar score. Note that this P-value depends
only the raw score -- it doesn't take into account either the length of the
query sequence or the total number of query sequences. The P-value is
estimated by fitting a normal distribution to the scores of the non-beta-helix
sequences in a non-redundant version of the PDB. You can think of it as a more
meaningful re-scaling of the raw score. P-values less than about 0.01 are
worth a second look. In our experience, false positives with very good scores
generally have a detectable sequence repeat; examples include a few of the
leucine rich repeat and hexapeptide repeat proteins. You have the option of
searching for these classes of proteins (see below).
Energy score:
SCWRL produces an energy score, which is a measure of how well the sidechains fit onto the given backbone. A high energy score (over 200 or 300) may be an indication that the wrap fits poorly onto the backbone, implying that the sequence may not form the motif. It may also indicate that, although the protein does fold into the specific motif, the sequence-structure alignment is inaccurate.
Reading a wrap:
BetaWrapPro provides several ways to view a wrap. One can download the PDB file provided, which contains the sequence-structure alignment in the 3D structure prediction.
Both the beta-helix and beta-trefoil wraps are presented as the entire fasta
sequence, and the PSIPRED secondary structure prediction, with the predicted
supersecondary structure highlighted.
In addition, the beta-helix wraps are shown in a stacking view, which highlights how the predicted rungs align. It should be noted that the "." characters in this view do not indicate gaps in the alignment, but are solely to keep the beta strands aligned correctly.
A key to the colors used for each motif is displayed at the bottom of the results page.
Wrap description:
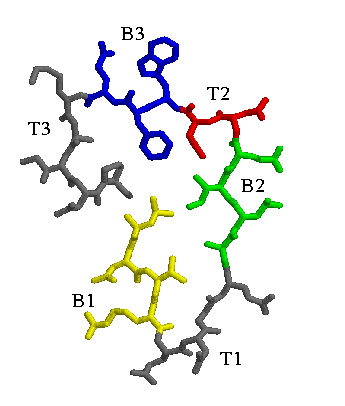
Beta-helix wraps are made up of five rungs, each consisting of three beta strands separated by turn regions.
Two of the turns can be highly variable, but the turn between strands two and
three is nearly always two residues long. A cross-section of a rung (from the PDB structure 1pcl, Pectate Lyase C) is shown here, with the turns and strands labeled.
Pfam score:
See below.
Advanced Options
At the bottom of the search form are some additional options which may improve
the speed of your search or filter out common false positive folds.
Motif options
You may choose to search for either one or both of the template motifs. If
you are only interested in one of them, it will be slightly faster to select
that fold rather than both.
Additional searches
BetaWrapPro is occasionally fooled by
sequences with simple sequence repeats that have similar structure to the
beta-helix motif: the hexapeptide repeat family and the leucine rich repeat
family. Pfam has generated profile HMMs for both of these families, and we
provide the option of searching for these repeats.
The Pfam and HMMER results attach an E-value to sequence hits. This E-value is an estimate of the expected number of hits with equal or better scores, given the number of query sequences. These E-values are estimated empirically by a calibration process involving random sequences.
Additional Assistance
If you encounter any problems running BetaWrapPro or have additional questions, please send mail to betawrap@csail.mit.edu.
